


EpiMapper
A Python package containing a full data analysis pipeline of ChIP-seq, CUT&RUN, ATAC-seq and CUT&Tag data.
Contents:
positional arguments: - task: Pipeline task to run
optional arguments: - -h, –help: Show this help message and exits
Usage: epimapper <task> [<args>]
Tasks available for using:
fastqc: Performs quality control on raw reads fastq files from high-thoughput sequencing.
bowtie2_alignment: Mapping reads to a reference genome with Bowtie2 alignment of fastq sequencing reads files from high-thoughput sequecing, and visulizing results.
remove_duplicates: Remove duplicated reads mapped to the same place in of a reference genome during alignment, and visulizing results.
fragment_length: Evaluation of mapped fragment length distribution of input SAM files exported from high-thoughput sequencing alignment, and visulizing results.
spike_in_calibration: Removes experimental bias by normalizing fragment counts based on sequencing depth to a spike-in genome and visulizes results.
peak_calling: Finds enriched regions/calls for peaks from chromatin profiling data with SEACR or MACS2, then visulizes results.
heatmap: Visualizes the enrichment of target protein in predefined genomic regions and peaks by creating heatmaps.
differential_analysis:Preforms differntial analysis on enriched reagions/peaks before annotating the stastitically significant changes to spesific genomic reagions and visulizing the results.
Files needed for a complete run of the pipeline:
Required:
FASTQ: Text files that contains the sequence data from next generation sequencing. The files must be stored togheter in one single directory.
Chromosome Sizes: Text file containin one column with information the chromosome sizes of the genome.
Genome Blacklist: A BED file conaining genome that should be avoided in the data analysis (i.e highly repetative regions)
Genome RefFlat Reference: A text (.txt) file containing the reference genome in RefFlat format.
either - Bowtie2 Indexing Files: Input file folder of Bowtie2 reference genome indexing files. or - Reference Genome FASTA: Input reference genome FASTA file for the creation of Bowtie2 reference genome.
Optional:
Enchancher: A sorted BED file containing the enhancer regions. This will be used as a refereence for annotation of the differntially bound marks.
Name requirements for files:
All input FASTQ files must follow a naming protocal, this is to ensure smooth transitions between the pipeline´s functions. The naming protocal is as following:
[SAMPEL-NAME]_rep[REPLICATION-NUMBER]_[R1/R2]_[(TECHNICAL-REPLICATE)].fastq
- The sampl name section must not contain underscore “_” or dot “.” in the name.
If there are none technical replicates, just skip the number.
Some examples:
H4K3me3_rep1_R2.fastq
ZNF143-Control-48h_rep1_R1.fastq
IgG_rep1_R2_001.fastq
Control-sample_rep2_R2.fastq
H4K27me3_rep1_R2_3.fastq
Any-thing-you-want-except-underscore-or-dot_rep2_R2.fastq
Output created by the pipeline
After a complete run of the EpiMapper pipeline you will be left with several newly created folders following the directory structure below.
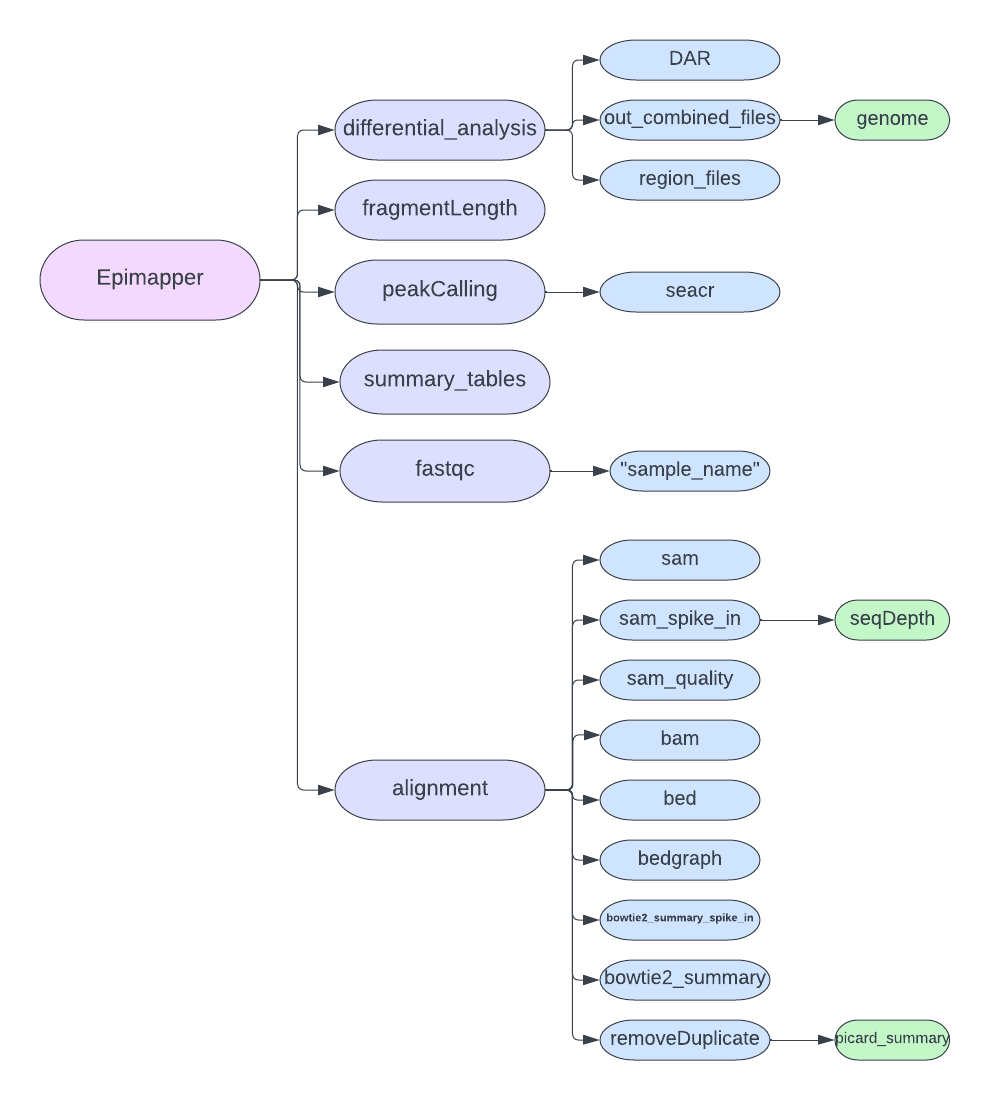
As the figure shows, and to make it eaiser for the user the main output directory “Epimapper” contains all the output from every subfunction. Please see each functions´documentation page to gain further knowledge about the output file of each repective function.